
Om Galafold®
VAD ÄR GALAFOLD®?
Galafold® (migalastat) är en den första och enda orala farmakologiska chaperon1 som är godkänd för långtidsbehandling av vuxna och ungdomar som är 12 år eller äldre med en bekräftad diagnos av Fabrys sjukdom och som har en behandlingsbar mutation.2
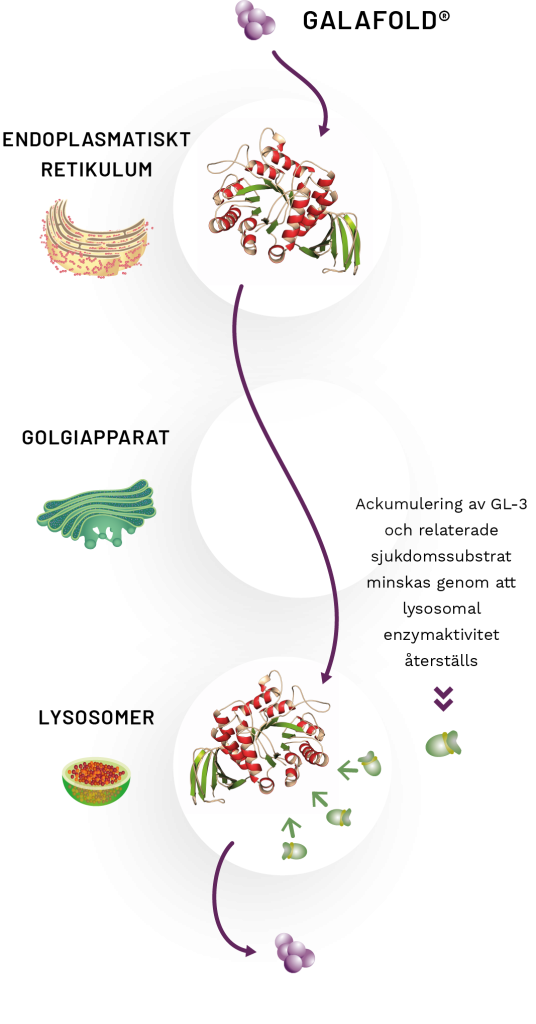
HUR FUNGERAR DET?
Galafold® är en första och enda1 orala farmakologiska chaperon som selektivt och reversibelt binder till kroppens eget försvagade enzym, stabiliserar det och underlättar dess korrekta transport till lysosomen.1 När Galafold® har nått lysosomen återställer separationen av Galafold® lysosomalaktiviteten hos enzymet α-galaktosidas A (α-Gal A), vilket leder till en minskning av sjukdomssubstraterna.2
Galafolds® föreslagna verkningsmekanism visas i bilden och kan sammanfattas som:
- Det stabiliserade α-Gal A-enzymet med bundet Galafold® lämnar det endoplasmatiska retikulum (ER)3
- När det stabiliserade enzymet med bundet Galafold når lysosomen separerar Galafold® från α-Gal A-enzymet2
- Det frisatta enzymet kan bryta ner GL-3 och relaterade sjukdomssubstrat2
- Lagrade substrat tros tränga undan Galafold® och ta över stabiliseringen av det muterade enzymet4
- Galafold® lämnar lysosomen och avsöndras från kroppen via urinen2
Galafold® har visat sig ha klinisk effekt och fått marknadsgodkännande baserat på två randomiserade fas 3-studier och oblindade förlängningsstudier1,5,6
Behandlingsbarhet
Galafold® är indicerat för långvarig behandling av vuxna och ungdomar från 12 års ålder som har en bekräftad diagnos på Fabrys sjukdom (α-galaktosidas A-brist) och som har en behandlingsbar mutation.2 Eftersom ett betydande antal mutationer hittills har identifierats2 är det väsentligt att mottagliga mutationer kan identifieras exakt, så att relevanta patienter med bekräftad diagnos av Fabrys sjukdom kan behandlas. Identifieringen av vilka GLA-mutationer som är behandlingsbara med Galafold® görs med Galafolds mottaglighetsanalys.
Mottagliga/behandlingsbara (amenable) GLA-mutationer är sådana som förväntas resultera i att α-Gal A-enzym kan bindas och stabiliseras av Galafold®, vilket innebär att α-Gal A a-aktiviteten överstiger förhandsspecificerade kriterier som är in vitro uppmätta genom en särskild analys som görs för att säkerställa att mutationen är mottaglig/behandlingsbar (amenable) för Galafold®.6 Denna analys som utförs av ett laboratorium, är inte avsedd att klassificera mutationer som patogena eller underlätta diagnostiseringen av Fabrys sjukdom.6 Se denna video för att få veta mer om analysen hur man identifierar mottagliga/behandlingsbara (amenabble) mutationer för Galafold®.
Validering av farmakogenetik för identifiering av Fabry-patienter som ska behandlas med migalastat

Definerade kriterier
För mottaglighet/behandlingsbarhet (amenability) krävs både en absolut ökning av α-Gal A-aktiviteten med ≥3 % av vildtyp och en relativ ≥1,2-faldig ökning över baslinjen.
Webbplatsen för Galafold®-mottaglighet innehåller en sökningsbar databas med GLA-mutationer som har analyserats för mottaglighet fram till i dag. Du hittar den på www.GalafoldAmenabilityTable.com.
Uppskattningsvis har 35–50 % av patienterna GLA-mutationer som är behandlingsbara med Galafold®.1
Utvecklingen av Galafolds GLP-validerade mottaglighetsanalys
| Mätningen av enzymaktiviteten Både absoluta och relativa ökningar av α-Gal A-aktiviteten analyseras. | För att vara behandlingsbar måste en mutation uppfylla kriterierna för både absoluta och relativa ökningar av α-Gal A-aktiviteten. Detta är utformat för att säkerställa både kliniskt relevanta (absolut ökning) och tillräckligt stabila (relativ ökning) ökningar av enzymaktiviteten.6 |
| Definierade mottaglighetskriterier För behandlingsbarhet krävs både en absolut ökning med ≥3 % av vildtyp och en relativ ≥1,2-faldig ökning över baslinjen | Potentiella kriterier jämfördes med en in vivo-analys av män för att fastställa vilka mottaglighetskriterier som bäst kan förutsäga klinisk respons. De valda kriterierna hade det högsta positiva förutsägbara värdet på 1,0 och ett negativt förutsägbart värde på 0,875. Känsligheten och noggrannheten för behandlingsbarhet är måttligt lägre hos kvinnor, vilket reflekterar svårigheten i att mäta det muterade enzymets respons i närvaro av vildtyp-α-Gal A från den andra GLA-allelen. Detta indikerar dock inte nödvändigtvis en lägre potential för klinisk nytta.6 |
| Migalastat-koncentration använd i analys En koncentration på 10 µmol/L migalastat valdes | In vitro-koncentrationen av migalastat på 10 µmol/L motsvarar den ungefärliga genomsnittliga maximala plasmakoncentrationen (Cmax) hos patienter med Fabrys sjukdom efter en enda oral dos på 150 mg migalastat hydroklorid.6 |
| Analys validerad Analysen validerades med data från kliniska fas 2- och fas 3-studier. | Mottaglighetskriterierna bekräftades ytterligare baserat på farmakodynamisk respons på Galafold® i fas 2- och fas 3-studier. Jämförelsen visade hög noggrannhet och känslighet, samt positiva och negativa förutsägbara värden.6 |
| Ytterligare kategoriserade mutationer Mutationer hos patienter i kliniska studier är representativa för alla mutationer som är behandlingsbara för Galafold®. | Mottagliga mutationer (n=51) från patienter i kliniska prövningar jämfördes med alla 348 kända mottagliga mutationer som var kända 2018.7 De 51 mottagliga mutationerna som ingår i kliniska studier är representativa för den större gruppen av alla identifierade behandlingsbara mutationer.7 |
| Webbplats för fastställande av mottaglighet På webbplatsen www.GalafoldAmenabilityTable.com kan mottagligheten fastställas | Galafolds® webbplats för mottaglighet innehåller en sökbar databas med GLA-mutationer som fram till idag har analyserats för mottaglighet/behandlingsbarhet kan användas till att avgöra om en viss patients mutation är behandlingsbar med Galafold®.2 |
Information om Galafolds mottaglighetsanalys
Inga blodprov eller DNA-prov krävs6
Testningen av varje mutation upprepas med 20-faldig redundans6
Referenser
- Hughes DA, Nicholls K, Shankar SP, et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet. 2017;54(4):288-296.
- Sammanfattning av produktens egenskaper. Galafold® 123 mg hård kapsel: https://www.fass.se/LIF/product?userType=0&nplId=20150626000015
- Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30.
- Sánchez-Fernández EM, García Fernández JM, Ortiz Mellet C. Glycomimetic-based pharmacological chaperones for lysosomal storage disorders: lessons from Gaucher, GM1-gangliosidosis and Fabry diseases. Chem Commun. 2016;52(32):5497-5515.
- Germain DP, Hughes DA, Nicholls K, et al. Treatment of Fabry’s disease with the pharmacologic chaperone migalastat. N Engl J Med. 2016;375(6):545-555.
- Benjamin ER, Della Valle MC, Wu X, et al. Validering av farmakogenetik för identifiering av Fabry-patienter som ska behandlas med migalastat. Genet Med. 2017;19(4):430-438 (and Supplementary Appendix).
- Wu X, Williams H, Bond S, Bronfin B, Benjamin ER. Migalastat amenability assay comparison between amenable mutations studied in clinical trials and the broader population of all amenable mutations. Presenterad vid 2018 American College of Medical Genetics and Genomics (ACMG) Clinical Genetics Meeting; April 10-14, 2018; Charlotte, NC.